LAPORAN PRAKTIKUM PATOGEN TUMBUHAN: ANALISIS VIRUS RGSV DAN R DENGAN MENGGUNAKAN PCR
LAPORAN PRAKTIKUM PATOGEN TUMBUHAN
ANALISIS VIRUS RGSV DAN R DENGAN MENGGUNAKAN PCR

Oleh:
Miftachurohman
12/334974/PN/12969
Asisten:
Destania Putri
Erwin Najamuddin
Niken R Paramita
Rusmi S. W.
LABORATORIUM PENYAKIT TERPADU
JURUSAN HAMA DAN PENYAKIT TUMBUHAN
FAKULTAS PERTANIAN
UNIVERSITAS GADJAH MADA
YOGYAKARTA
2014
TUJUAN
Mengetahui teknik analisis molekuler dengan menggunakan PCR
TINJAUAN PUSTAKA
Penyakit tungro adalah salah satu penyakit penting yang menyebabkan kehilangan hasil pada tanaman padi di beberapa negara Asia Tenggara, termasuk Indonesia. Penyakit ini disebabkan oleh dua jenis virus tungro, yaitu rice ragged stunt virus (RRSV) dan rice grassy stunt virus (RGSV). Kedua virus tersebut ditularkan oleh wereng colat, Nilaparvata lugens(Chettanachi et al. 1987).
Kesulitan yang dihadapi di dalam mengidentifikasi RGSV dengan hanya berdasarkan pada gejala luar tanaman sakit adalah sukarnya mem bedakan gejala yang disebabkan oleh RRSV dengan dengan gejala yang disebabkan oleh virus lain pada tanaman padi(Frischmuth, 2002). Untuk mengatasi kesulitan tersebut berbagai pendekatan telah dikembangkan, misalnya implementasi bioteknologi berbasis biologi molekuler seperti teknik hibridisasi asam nuk leat dengan menggunakan metode perpanjangan rantai polimerasi (polymerase chain reaction, PCR) (Raga dkk2004).
Aplikasi PCR sangat membantu dalam pengelolaan penyakit tungro karena dapat digunakan untuk: diagnosis penyakit tungro, deteksi dini infeksi virus tungro dan keberadaan vektor yang infektif, identifikasi dan karakterisasi strain virus, deteksi munculnya strain virus tungro yang baru, karakterisasi ketahanan varietas terhadap virus tungro, dan perakitan varietas tahan penyakit tungro me lalui upaya pemuliaan konvensional dan rekayasa genetik, seperti tanaman transgenik tahan tungro (Praptana dan Yasin 2008).
Reaksi berantai polymerase (Polymerase Chain Reaction, PCR) adalah suatu metode enzimatis untuk amplifikasi DNA dengan cara in vitro. PCR ini pertama kali dikembangkan pada tahun 1985 oleh Kary B. Mullis. Amplifikas DNA pada PCR dapat dicapai bila menggunakan primer oligonukleotida yang disebut amplimers. Primer DNA suatu sekuens oligonukleotida pendek yang berfungsi mengawali sintesis rantai DNA. PCR memungkinkan dilakukannya pelipatgandaan suatu fragmen DNA. Umumnya primer yang digunakan pada PCR terdiri dari 20 -30 nukleotida. DNA template (cetakan) yaitu fragmen DNA yang akan dilipatgandakan dan berasal dari patogen yang terdapat dalam spesimen klinik. Enzim DNA polimerase merupakan enzim termostabil Taq dari bakteri termofilik Thermus aquaticus. Deoksiribonukleotida trifosfat (dNTP) menempel pada ujung 3’ primer ketika proses pemanjangan dan ion magnesium menstimulasi aktivasi polymerase(Yusuf, 2010)
(Polymerase Chain Reaction, PCR) adalah suatu metode enzimatis untuk amplifikasi DNA dengan cara in vitro. Pada proses PCR diperlukan beberapa komponen utama, yaitu DNA cetakan, Oligonukleotida primer, Deoksiribonukelotida trifosfat (dNTP), Enzim DNA Polimerase, dan Komponen pendukung lain adalah senyawa buffer. Pada proses PCR menggunakan menggunakan alat termosiklus. Sebuah mesin yang memiliki kemampuan untuk memanaskan sekaligus mendinginkan tabung reaksi dan mengatur temperatur untuk tiap tahapan reaksi. Ada tiga tahapan penting dalam proses PCR yang selalu terulang dalam 30-40 siklus dan berlangsung dengn cepat yaitu denaturasi, anneling, dan pemanjangan untai DNA. Produk PCR dapat diidentifikasi melalui ukurannya dengan menggunakan elektroforesis gel agarosa. Teknik PCR dapat dimodifikasi ke dalam beberapa jenis diantaranya : PCR- RFLP, PCR – RAPD, nested- PCR,QuantitativePCR, RT- PCR dan inverse – PCR. Keunggulan PCR dikatakan sangat tinggi. Hal ini didasarkan atas spesifitas, efisiensi dan keakuratannya(Yuwono dan Tribowo, 2006).
METODE PRAKTIKUM
Praktikum Patogen Tumbuhan Acara 5 dengan judul Deteksi Molekular RNA Virus pada Vektor dilaksanakan pada hari Jumat sebanyak dua kali pengamatan yaitu pada tanggal, 14 dan 21 November 2014 di laboratorium integrated of plant diseases , Jurusan Hama dan Penyakit Tumbuhan, Fakultas Pertanian, Universitas Gadjah Mada. Alat yang digunakan dalam praktikum ini diantaranya, tube 1.5 ml, tube micropastle, tube filtercolumn, vortex, mesin sentrifugasi, micro pipet, sarung tangan, PCR, mesin elektroforesis, uv transluminator. Adapun bahan yang digunakan adalah wereng coklat, RB buffer, merkapto etanol, etanol, buffer W1, wash buffer, RNAse free water, kapataq extra hot start, kapatq buffer, MgCl2, dNTP, primer (F), Primer (R), taq polymerase, air, CDNA, gel agarose 1.5%, air 3.5 mikroliter, 5x reaction buffer 2 mikroliter, ribolock 0.5 mikroliter, 10 mM dNTP mix 1 mikroliter, rever aid 0.5 mikroliter, oligo primer 0.5 mikroliter, RNA template 2 mikroliter.
Praktikum dilakukan dalam empat tahap, yaitu ekstraksi wereng (RNA virus), RT PCR, PCR dan elektroforesis. Pada tahap ekstraksi wereng (RNA virus) dilakukan empat tahap inti, yaitu: 1) Lisis sel yang terdiri dari penggerusan 3 ekor wereng sebanyak 25 gr menggunakan 400 mikroliter RB buffer dan 4/8 mikroliter beta-mercaptoetanol di dalam tube 1.5 mikroliter menggunakan mikropastle lalu divortek. Hasil vortek diinkubasi pada suhu ruang selama 3 menit. Lalu supernatan dipindahkan ke filter column sebanyak 200-250 mikroliter. Kemudian disentrifuge 1000g selama 30 detik. Setelah itu supernatan dipindah hati-hati kedalam tube 1.5ml (jangan sampai ada endapan yang terikut). 2)Presipitasi sel yang terdiri dari hasil dari tahap lisis sel dengan 400 mikroliter etanol 70% steril divortek untuk dihomogenkan. Hasilnya dipindahkan ke RB column kemudian di sentrifuge 9690g selama 2 menit. Supernatan dibuang. 3) Pencucian adalah tahap untuk menghilangkan sisa etanol, DNA dan protein yang terdiri dari 400 mikroliter buffer W1 disentifuge 9690 selama 1 menit. Supernatan dibuang ditambah 600 mikroliter wash buffer yang disentrifuge 9690g selama 1 menit. Tahap ini dilakukan sebanyak 2 kali. Kemudian supernatan dibuang lalu di dry sentrifuge 9690g selama 5 menit. 4) RNA Elution yang terdiri dari RB column dipindahkan ke tube 1.5 ml ditambah 30 mikroliter RNAse freewater. Kemudian didiamkan selama 2 menit lalu disentrifuge 9690 selama 2 menit. Lalu didapatkanlah tube dengan RNA total wereng. Setelah tahap ekstraksi wereng masuk ke tahap RT PCR yaitu tahap reverse transkiptase dari RNA menjadi DNA. Kemudian dilakukanlah PCR.
Siklus PCR terdiri dari denaturasi awal pada suhu 94oC selama 3 menit. Pada saat ini, molekul DNA cetakan mengalami denaturasi sehingga kedua untaiannya terpisah. Pemisahan untaian ini diperlukan agar primer dapat menempel. Setelah beberapa menit pada suhu denaturasi, suhu alat diturunkan sehingga mencapai suhu yang sesuai untuk penempelan primer pada DNA cetakan. Suhu yang digunakan pada siklus ini adalah 53oC. Proses penempelan primer memerlukan waktu 1 menit, selanjutnya suhu alat dinaikkan ke suhu yang optimum yaitu 72oC untuk aktivitas polimerisasi DNA. Pada suhu inilah terjadi proses polimerisasi (sintesis) DNA baru dengan adanya aktivitas DNA polimerase, dNTP, primer dan DNA cetakan. Proses ini berlangsung selama 2 menit. Setelah proses polimerisasi, kemudian dilakukan lagi siklus seperti semula, yaitu dengan menaikkan suhu menjadi 94oC (denaturasi), kemudian diturunkan menjadi 53oC (penempelan primer), dan kemudian dinaikkan lagi menjadi 72oC (polimerisasi). Siklus perubahan suhu ini dilakukan berulang-ulang sekitar 30-40 kali. Hasil PCR kemudian dianalisis menggunakan elektroforesis gel agarose.
HASIL DAN PEMBAHASAN
HASIL
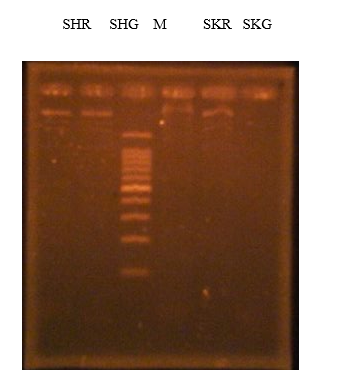
Keterangan
M : Marker (100 Bp)
SHR : wereng sehat dengan primer RRSV
SHG : Wereng Sehat dengen Primer RGSV
SKR : Wereng sakit dengan primer RRSV
SKG : Wereng sakit dengan Market RGSV
PEMBAHASAN
PCR adalah singkatan dari Polymerase Chain Reaction. Teknik ini merupakan teknik perbanyakan DNA secara in vitro. Teknik ini memungkinkan adanya amplifikasi antara dua region DNA yang diketahui, hanya di dalam tabung reaksi, tanpa perlu memasukkannya ke dalam sel (in vivo). Dalam sistem kerjanya, PCR dilandasi oleh struktur DNA. Dalam keadaan nativenya, DNA merupakan double helix, yang terdiri dari dua buah pita yangberpasangan antiparalel antara satu dengan yang lain dan berikatan dengan ikatan hidrogen. Ikatanhidrogen terbentuk antara basa-basa yang komplementer, yaitu antara basa Adenin (A) dengan Thymine (T), dan Guanine (G) dengan Cytosin (C). Basa-basa itu terikat dengan molekul gula, deoksiribosa, dan setiap satu molekul gula berikatan dengan molekul gula melalui ikatan fosfat.
Terdapat tiga tahap utama di dalam setiap siklusnya, yaitu :
- Denaturasi: Selama proses denaturasi, double strandedDNA akan membuka menjadi single strandedDNA. Hal ini disebabkan karenasuhu denaturasi yang tinggi menyebabkan putusnya ikatan hidrogen diantara basa-basa yang komplemen. Pada tahap ini, seluruh reaksi enzim tidak berjalan, misalnya reaksi polimerisasi pada siklus yang sebelumnya.
- Annealing: Primer akan menuju daerah yang spesifik, dimana daerah tersebut memiliki komplemen dengan primernya. Pada proses annealing ini, ikatan hidrogen akan terbentuk. Selanjutnya, DNA polymerase akanberikatan sehingga ikatan hidrogen tersebut akan menjadi sangat kuat dan tidak akan putus kembali apabila dilakukan reaksi polimerisasi selanjutnya, misalnya pada 72oC.
- Reaksi polimerisasi (extension): Umumnya, reaksi polimerisasi atau perpanjangan rantai ini, terjadi pada suhu 72oC. Primer yang telah menempel tadi akan mengalami perpanjangan dengan dNTP yang komplemen pada sisi 3’nya.
Komponen – komponen dalam reaction mixture PCR yaitu H2O steril, fungsinya sebagai pelarut campuran. Bufer berfungsi untuk mengkondisikan reaksi agar berjalan optimum dan menstabilkan enzim DNA polymerase. Bufer biasanya terdiri atas bahan-bahan kimia. Komponen lainnya yaitu dNTP (deoxynucleoside triphosphate) sebagai pembentuk basa komplementer dan penyusun DNA, terdiri atas 4 macam sesuai dengan basa penyusun DNA, yaitu dATP, dCTP, dGTP dan dTTP. Primer berfungsi untuk menginisiasi sintesis DNA pada sekuens target yang spesifik dan membatasi reaksi polimerisasi DNA. Primer terdiri dari dua macam, yaitu primer forward dan primer reverse. Primer forward untuk menginisiasi sintesis untai DNA dari ujung 5’ ke ujung 3’, sedangkan primer reverse menginisiasi sintesis DNA dari ujung 3’ ke ujung 5’. Kation divalen terdiri dari ion logam bivalen (umumnya Mg2+) dan ion logam monovalen (K+), berfungsi sebagai kofaktor bagi enzim DNA polymerase. Tanpa ion-ion tersebut enzim DNA polymerase tidak dapat bekerja. DNA template adalah DNA yang memiliki sekuens target untuk penempelan primer, berfungsi sebagai cetakan DNA yang akan diamplifikasi. Komponen yang terakhir yaitu enzim DNA polymerase berfungsi untuk membaca kode DNA serta menghubungkan pasangan nukleotida dalam menghasilkan salinan DNA.
Untuk mendapatkan hasil PCR yang optimal perlu dilakukan optimasi proses PCR. Secara umum optimasi proses PCR dapat dilakukan dengan cara memvariasikan kondisi yang digunakan pada proses PCR tersebut. Optimasi kondisi berkaitan erat dengan faktor-faktor seperti jenis polimerase DNA; suhu; konsentrasi, dalam hal ini berkaitan dengan dNTPs, MgCl2 dan DNA polimerase; buffer PCR dan waktu.
- Jenis polimerase DNA: Kemampuan mengkatalisis reaksi polimerasi DNA pada proses PCR yang terjadi pada tahap ekstensi untuk DNA rantai panjang akan berbeda dengan untuk DNA rantai pendek. Penggunaan jenis DNA polimerase tergantung pada panjang DNA target yang akan diamplifikasi. Untuk panjang fragmen DNA lebih besar dari tiga kilobasa akan memerlukan jenis polimerase dengan aktivitas tinggi.
- Konsentrasi dNTPs, MgCl2, polimerase DNA: Konsentrasi optimal dNTPs ditentukan oleh panjang target DNA yang diamplifikasi. Untuk panjang target DNA kurang dari satu kilobasa biasanya digunakan konsentrasi dNTPs sebanyak 100 uM, sedangkan untuk panjang target DNA lebih besar dari satu kilobasa diperlukan konsentrasi dNTPs sebanyak 200 uM. Umumnya konsentrasi optimal MgCl2 berkisar antara 1,0 – 1,5 mM. Konsentrasi MgCl2 yang terlalu rendah akan menurunkan perolehan PCR. Sedangkan konsentrasi yang terlalu tinggi akan menyebabkan akumulasi produk non target yang disebabkan oleh terjadinya mispriming. Jumlah polimerase DNA yang digunakan tergantung pada panjang fragmen DNA yang akan diamplifikasi. Untuk panjang fragmen DNA kurang dari dua kilobasa diperlukan 1,25 – 2 unit per 50 uL campuran reaksi, sedangkan untuk panjang fragmen DNA lebih besar dari dua kilobasa diperlukan 3 – unit per 50 uL campuran reaksi.
- Suhu: Pemilihan suhu pada proses PCR sangat penting karena suhu merupakan salah satu faktor yang menentukan keberhasilan suatu PCR. Dalam hal ini suhu berkaitan dengan proses denaturasi DNA templat, annealingdan ekstensi primer. Suhu denaturasi DNA templat berkisar antara 93 – 95 o C, ini semua tergantung pada panjang DNA templat yang digunakan dan juga pada panjang fragmen DNA target. Suhu denaturasi yang terlalu tinggi akan menurunkan aktivitas polimerase DNA yang akan berdampak pada efisiensi PCR. Selain itu juga dapat merusak DNA templat, sedangkan suhu yang terlalu rendah dapat menyebabkan proses denaturasi DNA templat tidak sempurna. Pada umumnya suhu denaturasi yang digunakan adalah 94 o Secara umum suhu annealingyang digunakan berkisar antara 37 – 60 o Pemilihan suhu annealingberkaitan dengan Tm primer yang digunakan untuk proses PCR. Suhu annealingyang digunakan dapat dihitung berdasarkan (Tm – 5) o C sampai dengan (Tm + 5)o C. Dalam menentukan suhu annealingyang digunakan perlu diperhatikan adanya mispriming pada daerah target dan nontarget, dan keberhasilan suatu proses PCR akan ditentukan oleh eksperimen. Proses ekstensi primer pada proses PCR selalu dilakukan pada suhu 72 O C karena suhu tersebut merupakan suhu optimum polimerase DNA yang biasa digunakan untuk proses PCR.
- Buffer PCR: Buffer PCR yang digunakan berkaitan dengan pH dan kapasitas buffer nya. Dalam perdagangan ada dua jenis buffer PCR yaitu “Low-salt buffer” (pH 8,75 dan kapasitas buffer rendah) dan “High-salt buffer” (pH 9,2 dan kapasitas buffer tinggi). Umumnya buffer PCR tersedia sesuai dengan jenis polimerase DNA nya. Penggunaan jenis buffer ini tergantung pada DNA target yang akan diamplifikasi. Untuk panjang DNA target antara 0 – 5 kilobasa biasanya diperlukan “low-salt buffer” sedangkan untuk panjang DNA target lebih besar dari lima kilobasa digunakan “high-salt buffer”.
- Waktu: Pemilihan waktu yang digunakan berkaitan dengan proses denaturasi DNA templat, annealingdan ekstensi primer. Untuk denaturasi DNA templat umumnya dilakukan selama 30 – 90 detik, ini semua tergantung pada DNA templat yang digunakan. Waktu denaturasi yang terlalu lama akan merusak templat DNA dan sekaligus dapat menurunkan aktivitas polimerase DNA. Sedangkan waktu denaturasi yang terlalu pendek akan menyebabkan proses denaturasi tidak sempurna. Penentuan waktu untuk proses annealing berkaitan dengan panjang primer. Untuk panjang primer 18 – 22 basa cukup dengan 30 detik, sedangkan untuk panjang primer lebih besar dari 22 basa diperlukan waktu annealing 60 detik. Pemilihan waktu ekstensi primer tergantung pada panjang fragmen DNA yang akan diamplifikasi. Secara umum untuk mengamplifikasi setiap satu kilo basa DNA diperlukan waktu 30 – 60 detik. Pada setiap melakukan PCR harus dilakukan juga kontrol positif, ini diperlukan untuk memudahkan pemecahan masalah apabila terjadi hal yang tidak diinginkan. Selain itu juga harus dilakukan terhadap kontrol negative untuk menghindari kesalahan positif semu.
Teknik PCR memiliki kelebihan dan kekurangan. Kelebihan dari teknik PCR untuk deteksi molekuler adalah antara lain:
- Memiliki spesifisitas tinggi.
- Sangat cepat, dapat memberikan hasil yang sama pada hari yang sama.
- Dapat membedakan varian mikroorganisme.
- Mikroorganisme yang dideteksi tidak harus hidup.
- Mudah di set up.
Adapun kekurangan dari teknik PCR adalah anatra lain:
- Sangat mudah terkontaminasi.
- Biaya peralatan dan reagen mahal.
- Interpretasi hasil PCR yang positif belum tervalidasi untuk semua penyakit infeksi (misalnya infeksi pasif atau laten).
- Teknik prosedur yang kompleks dan bertahap membutuhkan keahlian khusus untuk melakukannya.
Hasil praktikum ini menunjukkan bahwa pita fragmen DNA yang disinari oleh sinar UV adalah negative karena tidak menunjukkan adanya pergerakan. Pergerakan yang terjadi merupakan pergerakan dari marker DNA pada tepi sel agarose. Jika pita fragmen ini menunjukkan hasil yang positif, maka fragmen DNA RRSV dan RGSV yang terdeteksi akan mencapai migrasi hingga 500bp, dimana fragmen ini merupakan fragmen paling terang yang terdapat pada marker. Ada beberapa hal yang dapat menyebabkan keadaan ini tejadi, diantaranya adlalah karena penyimpanan ekstraksi DNA yang terlalu lama setelah diisolasi pada praktikum sebelumnya. Selain itu, pada saat menuangkan ektraksi DNA ke sumuran terjadi kesalahan, yaitu penuangan tidak sepenuhnya tertuang ke sumuran. Bahan yang digunakan untuk PCR juga dapat menyebabkan hasil PCR menjadi error, karena terlalu lama penimpanan ataupun terkontaminasi. Selain itu, wereng yang di ekstrak juga mungkin steril, sehingga tidak ditemukan DNA virus target, yaitu virus tungro.
KESIMPULAN
PCR adalah teknologi canggih yang dapat mendeteksi DNA dengan cara amplifikasi DNA. Hasil pemeriksaan PCR dapat membantu untuk menegakkan diagnosa sepanjang pemeriksaan tersebut dikerjakan dengan cara yang benar dan sesuai dengan standar internasional. Keunggulan PCR dikatakan sangat tinggi. Hal ini didasarkan atas spesifitas, efisiensi dan keakuratannya. Masalah yang berkenaan dengan PCR yaitu biaya PCR yang masih tergolong tinggi
DAFTAR PUSTAKA
Chettanachit, D., W. Rattanakarn, and J. Hongkajorn. 1987. Studies of factors causing variation of varietal reaction to yellow orange leaf virus. Annual Report of Division of Plant Pathology and Microbiology, Deparlment of Agriculture, Bangkok.pp.78-87
Frischmuth, T. 2002. Plant Viruses AS Molecular Pathogens. The Haworth Press Inc, USA.
Praptana, R.H. dan M. Yasin. 2008. Peranan bioteknologi dalam pengelolaan penyakit tungro. Iptek Tanaman Pangan. 3: 98-103.
Raga, I.N., W. Murdita, M.P.L. Tri, S.W. Edi, dan Oman, 2004. Sistem surveillance antisipasi ledakan penyakit tungro di Indonesia. Prosiding Seminar Nasional Status Program Penelitian Tungro Mendukung Keberlanjutan Produksi Padi Nasional. Makassar, 7-8 September 2004
Yusuf, Z.K. 2010. Polymerase Chain Reaction (PCR). Saintek 5: –
Yuwono dan Tribowo, 2006. Teori dan Aplikasi Polymerase Chain Reaction, Panduan Eksperimen PCR untuk Memecahkan Masalah Biologi Terkini, Penerbit Andi, Yogyakarta
